

Quantinuum introduces hybrid solver for industrially relevant chemical modeling
New solution could have significant impacts for the automotive, aerospace, semiconductor and oil and gas industries
It’s believed that quantum computing will transform the way we solve chemistry problems, and the Quantinuum scientific team continues to push the envelope towards making that a reality.
In their latest research paper published on the arXiv, Quantinuum scientists describe a new hybrid classical-quantum solver for chemistry. The method they developed can model complex molecules at a new level of efficiency and precision.
Dr. Michał Krompiec, Scientific Project Manager, and his colleague Dr. David Muñoz Ramo, Head of Quantum Chemistry, co-authored the paper, "Strongly Contracted N-Electron Valence State Perturbation Theory Using Reduced Density Matrices from a Quantum Computer".
The implications are significant as their innovation “tackles one of the biggest bottlenecks in modelling molecules on quantum computers,” according to Dr. Krompiec.
Quantum computers are a natural platform to solve chemistry problems. Chemical molecules are made of many interacting electrons, and quantum mechanics can describe the behavior and energies of these electrons.
As Dr. Krompiec explains, “nature is not classical, it is quantum. We want to map the quantum system of interacting electrons into a quantum system of interacting qubits, and then solve it.”
Solving the full picture of electron interactions is extremely difficult, but fortunately it is not always necessary. Scientists usually simplify the task by focusing on the active space of the molecule, a smaller subset of the problem which matters most.
Even with these simplifications, difficulties remain. One challenge is carefully choosing this smaller subset, which describes strongly correlated electrons and is therefore more complex. Another challenge is accurately solving the rest of the system. Solving the chemistry of the complex subset can often be done from perturbation theory using so-called “multi-reference” methods.
In their work, the Quantinuum team came up with a new multi-reference technique. They maintain that only the strongly correlated part of the molecule should be calculated on a quantum computer. This is important, as this part usually scales exponentially with the size of the molecule, making it classically intractable.
The quantum algorithm they used on this part relied on measuring reduced density matrices and feeding them into a multi-reference perturbation theory calculation, a combination that had never been used in this context. Implementing the quantum electronic structure solver on the active space and using measured reduced density matrices makes the problem less computationally expensive and the solution more accurate.
The team tested their workflow on two molecules - H2 and Li2 – using Quantinuum’s hybrid solver implemented in the InQuanto quantum computational chemistry platform and IBM’s 27-qubit device. Quantinuum software is platform inclusive and is often tested on both its own H Series ion-trap quantum systems as well as others.
The non-strongly correlated regions of the molecules were run classically, as they would not benefit from a quantum speedup. The team’s results showed excellent agreement with previous models, meaning their method worked. Beyond that, the method showed great promise for reaching new levels of speed and accuracy for larger molecules.
The future impact of this work could create a new paradigm to perform quantum chemistry. The authors of the paper believe it may represent the best way of computing dynamic correlation corrections to active space-type quantum methods.
As Dr. Krompiec said, “Quantum chemistry can finally be solved with an application of a quantum solver. This can remove the factorial scaling which limits the applicability of this rigorous method to a very small subsystem.”
The idea to use a multi-reference method along with reduced density matrix measurement is quite novel and stems from the diverse backgrounds of the team at Quantinuum. It is a unique application of well-known quantum algorithms to a set of theoretical quantum chemistry problems.
What’s Next
The use cases are vast. Analysis of catalyst and material properties may first benefit from this new method, which will have a tremendous impact in the automotive, aerospace, fine chemicals, semiconductor, and energy industries.
Implementing this method on real hardware is limited by the current noise levels. But as the quality of the qubits increases, the method will unleash its full potential. Quantinuum’s System Model H1 trapped-ion hardware, Powered by Honeywell, benefits from high fidelity qubits, and will be a valuable resource for quantum chemists wishing to follow this work.
This hybrid quantum-classical method promises a path to quantum advantage for important chemistry problems, as machines become more powerful.
As Dr. Krompiec summarizes, “we haven’t just created a toy model that works for near-term devices. This is a fundamental method that will still be relevant as quantum computers continue to mature.”
About Quantinuum
Quantinuum, the world’s largest integrated quantum company, pioneers powerful quantum computers and advanced software solutions. Quantinuum’s technology drives breakthroughs in materials discovery, cybersecurity, and next-gen quantum AI. With over 500 employees, including 370+ scientists and engineers, Quantinuum leads the quantum computing revolution across continents.
- Quantinuum continues its progress toward fault-tolerant quantum computing, with a series of peer-reviewed breakthroughs in fault-tolerant operations.
- Our progress is not only scientific; it is commercial. By improving logical-qubit reliability and encoding efficiency, Quantinuum is reducing the resource overhead required to scale its quantum computers toward commercially useful workloads.
- These results were achieved on commercial Quantinuum hardware, reinforcing that our architecture is not just setting new standards, but building a practical foundation for customers, partners, and researchers preparing for the fault-tolerant era.
Fault-tolerant quantum computing is the threshold the industry must cross before quantum computers can solve the hardest, highest-value problems with confidence. To be commercially useful at scale, the question is not simply who can build more qubits. It is who can build reliable, efficient, scalable systems that reduce technical risk and accelerate the path to commercial usefulness.
Quantinuum is progressing on that path.
Last year, in partnership with Microsoft, we published a breakthrough in logical computing, demonstrating logical qubits that outperformed their physical counterparts by a factor of 800. We are proud to announce that this work is now being published in Nature, one of the most highly regarded scientific journals in the world.
This work highlights our leading fidelities, as shown in Table 1:
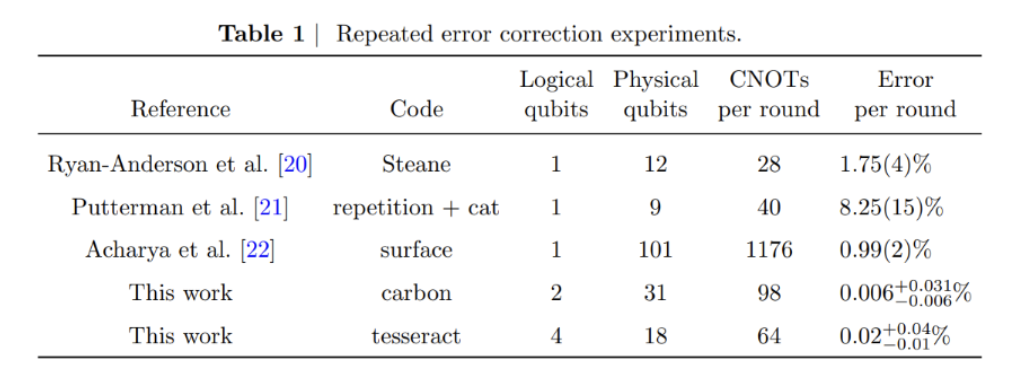
Since then, we’ve accelerated our efforts to reach large-scale fault tolerance and advanced what we believe to be the core building blocks of fault-tolerant quantum computing, from logical-qubit teleportation and multiple error-correction breakthroughs to one of the first meaningful computations using logical qubits. Importantly, these results were achieved on commercial Quantinuum hardware, demonstrating not just scientific progress, but a practical and efficient path toward scalable, customer-ready fault tolerance.
A Recap of Our Recent Technical Progress
Since the work with Microsoft, we achieved a milestone years ahead of schedule, demonstrating high-fidelity teleportation of a logical qubit, which was published in Science, one of the world’s most prestigious journals. Later, we beat our own record in this crucial fault tolerance milestone, thanks to continued improvements to our System Model H2’s fidelity.
Then, a series of results demonstrating more error-correcting milestones (and codes):
- Better than physical results in a high-rate non-local code,
- First-ever demonstration of a single-shot error correcting code (which significantly reduces resource requirements) in 4 dimensions
- Extending qubit lifetimes by 10x with a concatenated code
- Observed fault-tolerance thresholds with concatenated codes
- High fidelity magic states and a fully fault tolerant universal gate set in two different papers
Recently, we topped ourselves yet again by performing one of the first meaningful computations with logical qubits – exploring key questions in materials and magnetism, using logical qubits with better error rates than their physical counterparts. This result also includes a leading “encoding rate” squeezing 48 logical qubits out of just 98 physical qubits, emphasizing how our architecture helps to support large scale fault tolerance without enormous resource costs.
It is worth noting that all these results were achieved on our commercial hardware, not on one-off laboratory test-stands – reflecting the performance that we are able to deliver to our customers.
We also did crucial theoretical work, exploring new options for error correction that can reduce resource requirements, time to solution, and shorten the timeline to large scale fault tolerance.
Commercial Implications and the Road Ahead
We believe the commercial implication is clear: Quantinuum is reducing the uncertainty around the path to fault-tolerant quantum computing. Our architecture, hardware fidelity, full-stack control, and error-correction progress are converging into a practical roadmap for systems that can support valuable scientific and commercial workloads.
For those evaluating when quantum computing will become strategically relevant, we believe the signal is also increasingly clear: the fault-tolerant era is no longer a distant concept. It is becoming an engineering reality, and Quantinuum is leading the way.
- University of Southern Denmark (SDU) to use Quantinuum Helios, supported by the Danish e-Infrastructure Consortium (DeiC)
- Access to Helios enables SDU to test and refine fault-tolerant algorithms and error-correction codes under realistic hardware conditions
- The collaboration supports at a scale of 48 logical qubits, positioning Denmark at the forefront of scalable, practical quantum computing
- Researchers exploring the scientific foundations for future development of applications in fields including pharmaceuticals, finance, and defense
Progress in quantum computing is measured by hardware advances plus the algorithms and quantum error-correction codes that turn quantum systems into useful computational tools.
Thanks to recent hardware advances, researchers are increasingly sharpening their tools to probe the performance of quantum algorithms and understand how they behave in realistic conditions – where stability, system architecture and algorithm design all shape performance.
A new Denmark-based collaboration between the University of Southern Denmark (SDU), Quantinuum, and the Danish e-Infrastructure Consortium (DeiC) will utilize Quantinuum Helios. Researchers at the SDU’s Centre for Quantum Mathematics, led by Jørgen Ellegaard Andersen, will use Helios to pursue research into topological quantum computing.
Their work could help explain how and why successful quantum algorithms perform as they do, informing the development of high-performance algorithms suited to emerging quantum systems. They’re exploring the scientific foundations that support future quantum applications across areas including pharmaceuticals, finance, and defense.
“We are thrilled to gain access to Quantinuum’s high-fidelity Helios system. This collaboration gives us a unique opportunity to test the limits of our algorithms and evaluate system performance, while advancing fundamental research and laying the foundation for future applications.”
— Professor Jørgen Ellegaard Andersen, Director of the Centre for Quantum Mathematics at University of Southern Denmark
Why topological methods matter
Topological quantum computing is an area of research that connects quantum computation with deep mathematical structures. It includes the study of error correcting codes known as surface codes that encode quantum information in the global properties of systems of logical qubits.
The research team will explore how these codes behave, and how they may support the development of fault-tolerant quantum algorithms in practical implementations under realistic conditions.
This distinction between theory and practical implementation matters. In theory, topological approaches offer a rich framework for designing algorithms and error-correcting codes. In practice, researchers need to understand how those ideas perform when implemented on real systems, where questions of noise, stability, overhead, and scaling become central. The collaboration will allow the SDU team to investigate these questions directly.
New ways to benchmark quantum processors
Beyond individual algorithms and codes, the research will also develop tools for benchmarking quantum processors. The goal is to develop new ways to characterize fidelity and stability in regimes that can be difficult to access.
The team will also explore hybrid quantum–classical approaches, including machine-learning techniques assisted by quantum hardware, to study the mathematical structures at the heart of topological quantum computing. This work reflects a broader field of research in which quantum and classical methods are used together, each contributing to parts of a computational problem.
Strengthening Denmark’s quantum ecosystem
The collaboration reflects the growing role of national quantum infrastructure in supporting research and talent development. Denmark has a long tradition of scientific innovation, and this collaboration is intended to support the country’s continued development in quantum technology.
The initiative is supported by DeiC, which played a central role in securing funding and enabling access to Quantinuum’s systems. DeiC has been assigned a particular role in developing and coordinating quantum infrastructure initiatives for the benefit of universities and industry, operating without its own commercial, sectoral, or geographical interests. This includes securing dedicated access to quantum computers, producing advisory services and supporting the development of new talent in the Danish quantum sector.
“DeiC’s special effort to secure funding and access for this research initiative is rooted in our organization’s role in relation to the Danish Government’s strategy for quantum technology.”
— Henrik Navntoft Sønderskov, Head of Quantum at Danish e-Infrastructure Consortium
This collaboration promises to accelerate the development of practical algorithms. It is grounded in fundamental science – but its focus is practical: discovering and testing mathematical approaches to topological quantum computing that can be implemented, evaluated, and improved on real quantum hardware.
That work requires both theoretical insight and access to a system such as Helios capable of supporting meaningful scientific work.

This month, Quantinuum welcomed its global user community to the first-ever Q-Net Connect, an annual forum designed to spark collaboration, share insights, and accelerate innovation across our full-stack quantum computing platforms. Over two days, users came together not only to learn from one another, but to build the relationships and momentum that we believe will help define the next chapter of quantum computing.
Q-Net Connect 2026 drew over 170 attendees from around the world to Denver, Colorado, including representatives from commercial enterprises and startups, academia and research institutions, and the public sector and non-profits - all users of Quantinuum systems.
The program was packed with inspiring keynotes, technical tracks, and customer presentations. Attendees heard from leaders at Quantinuum, as well as our partners at NVIDIA, JPMorganChase and BlueQubit; professors from the University of New Mexico, the University of Nottingham and Harvard University; national labs, including NIST, Oak Ridge National Laboratory, Sandia National Laboratories and Los Alamos National Laboratory; and other distinguished guests from across the global quantum ecosystem.
Congratulations to Q-Net Connect 2026 Award Recipients!
The mission of the Quantinuum Q-Net user community is to create a space for shared learning, collaboration and connection for those who adopt Quantinuum’s hardware, software and middleware platform. At this year’s Q-Net Connect, we awarded four organizations who made notable efforts to champion this effort.
- JPMorganChase received the ‘Guppy Adopter Award’ for their exemplary adoption of our quantum programming language, Guppy, in their research workflows.
- Phasecraft, a UK and US-based quantum algorithms startup, received the ‘Rising Star’ award for demonstrating exceptional early impact and advancing science using Quantinuum hardware, which they published in a December 2025 paper.
- Qedma, a quantum software startup, received the ‘Startup Partner Engagement’ award for their sustained engagement with Quantinuum platforms dating back to our first commercially deployed quantum computer, H1.
- Anna Dalmasso from the University of Nottingham received our ‘New Student Award’ for her impressive debut project on Quantinuum hardware and for delivering outstanding results as a new Q-Net student user.
Congratulations, again, and thank you to everyone who contributed to the success of the first Q-Net Connect!
Become a Q-Net Member
Q-Net offers year‑round support through user access, developer tools, documentation, trainings, webinars, and events. Members enjoy many exclusive benefits, including being the first to hear about exclusive content, publications and promotional offers.
By joining the community, you will be invited to exclusive gatherings to hear about the latest breakthroughs and connect with industry experts driving quantum innovation. Members also get access to Q‑Net Connect recordings and stay connected for future community updates.